Nel meccanismo di replicazione cellulare, esiste un sistema, chiamato checkpoint di fase S, programmato per il funzionamento ottimale della replicazione .In condizioni patologiche, come nel caso di tumori , il sistema presenta uno stato di "stress replicativo" cronico.
L'accumulo di mutazioni in grado di alterare la funzione di alcune delle proteine chiave del checkpoint di fase S, fa si' che i tumori siano caratterizzati da una crescente instabilita' genomica. Il team di ricercatori, coordinati da Pietro Pichierri e Annapaola Franchitto dell'ISS, in collaborazione con la Prof.ssa Spies dell'Universita' dell'Iowa negli Stati Uniti, hanno individuato le due proteine responsabili della gestione di uno stato di replicazione patologica.
L'indagine, supportata dall'AIRC e dall'AICR, è stata pubblicata su Plos Genetics.
"Nel nostro studio - spiega Pichierri - abbiamo scoperto che cellule con alterata funzione del checkpoint di fase S (da noi alterate tramite l'inibizione di una delle proteine chiave di questa via molecolare, la chinasi CHK1) danno il via ad un meccanismo, non attivato in cellule normali, che prevede la funzione di due enzimi, chiamati RAD52 e MUS81, per supportare la sopravvivenza cellulare in condizione di stress replicativo. Tuttavia, l'azione di RAD52 e MUS81, sebbene garantisca la sopravvivenza di cellule deficienti per il checkpoint, determina anche un accumulo di instabilita' cromosomica. Infatti, l'inibizione della via RAD52-MUS81 previene l'accumulo di instabilita' genomica sin dalle prime fasi di sviluppo del tumore limitando la capacita' di sviluppare ulteriori mutazioni". Da qui lo spiraglio per una nuova terapia. "I nostri risultati aprono la via a potenziali approcci terapeutici che limitino l'insorgenza dell'instabilita' genomica oppure sinergizzino con essa per determinare una morte selettiva delle cellule tumorali, facendo cioe' di queste vie molecolari un target terapeutico - conclude Pichierri - Inoltre, poiche' gli inibitori di CHK1 cominciano ad essere valutati in terapia antitumorale, il nostro studio suggerisce che la contestuale inibizione di CHK1 e di MUS81/RAD52 possa rappresentare una piu' potente target therapy e la terapia preferenziale in quei tumori dove, per la presenza di mutazioni di CHK1, la monoterapia con gli inibitori di CHK1 non possa essere applicata".
ENGLISH VERSION PLOS GENETICS
RESEARCH ARTICLE
Survival of the Replication Checkpoint Deficient Cells Requires MUS81-RAD52 Function
Ivana Murfuni,
Affiliation: Section of Experimental and Computational Carcinogenesis, Department of Environment and Primary Prevention, Istituto Superiore di Sanità, Rome, Italy
X
Giorgia Basile,
Affiliation: Section of Molecular Epidemiology, Department of Environment and Primary Prevention, Istituto Superiore di Sanità, Rome, Italy
X
Shyamal Subramanyam,
Affiliations: Department of Biochemistry, University of Illinois Urbana-Champaign, Urbana, Illinois, United States of America, Department of Biochemistry, Carver College of Medicine, University of Iowa, Iowa City, Iowa, United States of America
X
Eva Malacaria,
Affiliation: Section of Experimental and Computational Carcinogenesis, Department of Environment and Primary Prevention, Istituto Superiore di Sanità, Rome, Italy
X
Margherita Bignami,
Affiliation: Section of Experimental and Computational Carcinogenesis, Department of Environment and Primary Prevention, Istituto Superiore di Sanità, Rome, Italy
X
Maria Spies,
Affiliation: Department of Biochemistry, Carver College of Medicine, University of Iowa, Iowa City, Iowa, United States of America
X
Annapaola Franchitto mail,
* E-mail: annapaola.franchitto@iss.it (AF); pietro.pichierri@iss.it (PP)
Affiliation: Section of Molecular Epidemiology, Department of Environment and Primary Prevention, Istituto Superiore di Sanità, Rome, Italy
X
Pietro Pichierri mail
* E-mail: annapaola.franchitto@iss.it (AF); pietro.pichierri@iss.it (PP)
Affiliation: Section of Experimental and Computational Carcinogenesis, Department of Environment and Primary Prevention, Istituto Superiore di Sanità, Rome, Italy
X
Abstract
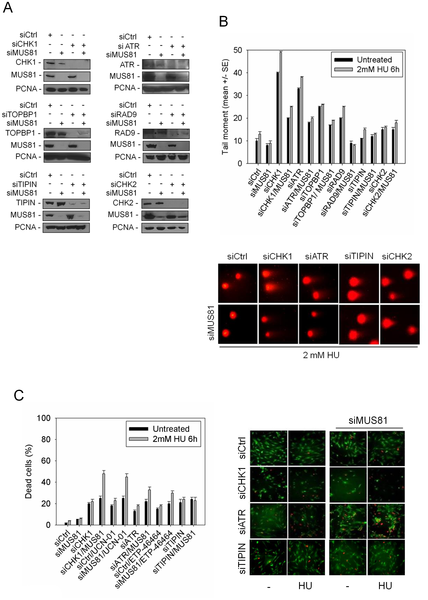
In checkpoint-deficient cells, DNA double-strand breaks (DSBs) are produced during replication by the structure-specific endonuclease MUS81. The mechanism underlying MUS81-dependent cleavage, and the effect on chromosome integrity and viability of checkpoint deficient cells is only partly understood, especially in human cells. Here, we show that MUS81-induced DSBs are specifically triggered by CHK1 inhibition in a manner that is unrelated to the loss of RAD51, and does not involve formation of a RAD51 substrate. Indeed, CHK1 deficiency results in the formation of a RAD52-dependent structure that is cleaved by MUS81. Moreover, in CHK1-deficient cells depletion of RAD52, but not of MUS81, rescues chromosome instability observed after replication fork stalling. However, when RAD52 is down-regulated, recovery from replication stress requires MUS81, and loss of both these proteins results in massive cell death that can be suppressed by RAD51 depletion. Our findings reveal a novel RAD52/MUS81-dependent mechanism that promotes cell viability and genome integrity in checkpoint-deficient cells, and disclose the involvement of MUS81 to multiple processes after replication stress.
Author Summary
The replication checkpoint ensures a smooth duplication of the genome. It counteracts the replication stress, which can cause chromosome rearrangements as found in most tumours. Given the importance of dealing with perturbed replication, and since in tumours secondary mutations or epigenetic changes may hamper efficiency of the replication checkpoint, it is crucial to determine the mechanisms responding to replication perturbation upon checkpoint inactivation. Furthermore, it is highly relevant to understand how failure of these mechanisms correlates with chromosomal damage after replication perturbation. Here, we investigated pathways that, in checkpoint-deficient human cells, are involved in the handling of perturbed DNA replication forks, and we uncovered a previously unappreciated function of RAD52 and MUS81 in ensuring viability of cells, but at the expense of genome instability. We also demonstrated that checkpoint deficiency can trigger different mechanisms of recovery from replication arrest depending on the presence of RAD52 or MUS81, resulting in a poor survival and reduced genome instability or increased survival and chromosomal damage. Our work provides new clues about how human cells deal with replication stress, and how genome instability may arise in cancer cells.
Citation: Murfuni I, Basile G, Subramanyam S, Malacaria E, Bignami M, et al. (2013) Survival of the Replication Checkpoint Deficient Cells Requires MUS81-RAD52 Function. PLoS Genet 9(10): e1003910. doi:10.1371/journal.pgen.1003910
Editor: Nancy Maizels, University of Washington, United States of America
Received: April 21, 2013; Accepted: September 11, 2013; Published: October 31, 2013
Copyright: © 2013 Murfuni et al. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
Funding: This work was supported by a grant from the Association for International Cancer Research (Grant #07-497) to PP, by grants from Associazione Italiana per la Ricerca sul Cancro (AIRC) to PP (IG #9294 and #13398) and AF (IG #11871) and by American Cancer Society grant RSG-09-182-01-DMC to MS. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Competing interests: The authors have declared that no competing interests exist.
LINK PLOS , FULL ARTICLE:
http://www.plosgenetics.org/article/info%3Adoi%2F10.1371%2Fjournal.pgen.1003910 |